Tutorial 4: Five modality integration
In this tutorial, we demonstrate how to use SCIGMA to analyze a Mouse Brain Spatial-Mux-seq dataset. This tutorial can be run in under 25 minutes on a GPU-enabled computer.
[1]:
%load_ext autoreload
%autoreload 2
[2]:
from SCIGMA_multi import *
from sklearn.neighbors import kneighbors_graph
from data import load_data, preprocessing, build_network, feature_graph, clr_normalize_each_cell
import numpy as np
import matplotlib.pyplot as plt
from utils import clustering, separation_scores, compactness_scores, lsi
import scanpy as sc
import gc
import pandas as pd
import anndata as ad
from scipy import sparse
Load data
To run this tutorial, make sure to download the Spatial Mux-seq dataset from here: - Epigenomics modalities: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM8494157 - RNA: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM8494158 - Protein: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM8494159 Modify the filepaths accordingly to match the downloaded data filepaths.
[ ]:
# Filepaths (modify)
sp_path = '<Downloaded data directory>'
file_RNA = "5M_20um_RNA/matrix.mtx.gz"
file_RNA_feat = "5M_20um_RNA/features.tsv.gz"
file_RNA_barcode = "5M_20um_RNA/barcodes.tsv.gz"
file_RNA_spatial = "5M_20um_spatial/tissue_positions_list.csv"
file_Protein = "5M_20um_Protein/matrix.mtx.gz"
file_Protein_feat = "5M_20um_Protein/features.tsv.gz"
file_Protein_barcode = "5M_20um_Protein/barcodes.tsv.gz"
file_Protein_spatial = "5M_20um_spatial/tissue_positions_list.csv"
file_ATAC = "GSM8494157_5M_20um_ATAC.fragments.tsv.gz"
file_HISTac = "GSM8494157_5M_20um_H3K27ac.fragments.tsv.gz"
file_HISTme3 = "GSM8494157_5M_20um_H3K27me3.fragments.tsv.gz"
file_protein = "GSM8494159_5M_20um_protein.tar.gz"
[ ]:
# RNA
rna = ad.read_mtx(sp_path+file_RNA).transpose()
rna_feat = pd.read_csv(sp_path+file_RNA_feat, sep='\t', header=None)
rna_barcode = pd.read_csv(sp_path+file_RNA_barcode, sep='\t', header=None)
rna_spatial = pd.read_csv(sp_path+file_RNA_spatial, sep=',', header=None)
rna.obs_names = rna_barcode[0]
rna.var_names = rna_feat[1]
rna.var['ensembl'] = list(rna_feat[0])
coord = pd.merge(rna_barcode, rna_spatial[[0,2,3]], left_on=0, right_on=0)
rna.obs['x'] = list(coord[2])
rna.obs['y'] = list(coord[3])
rna.obsm['spatial'] = rna.obs[['x','y']].to_numpy()
[ ]:
# Protein
protein = ad.read_mtx(sp_path+file_Protein).transpose()
protein_feat = pd.read_csv(sp_path+file_Protein_feat, sep='\t', header=None)
protein_barcode = pd.read_csv(sp_path+file_Protein_barcode, sep='\t', header=None)
protein_spatial = pd.read_csv(sp_path+file_Protein_spatial, sep=',', header=None)
protein.obs_names = protein_barcode[0]
protein.var_names = protein_feat[0]
protein.var['names'] = list(protein_feat[0])
coord = pd.merge(protein_barcode, protein_spatial[[0,2,3]], left_on=0, right_on=0)
protein.obs['x'] = list(coord[2])
protein.obs['y'] = list(coord[3])
protein.obsm['spatial'] = protein.obs[['x','y']].to_numpy()
[ ]:
# ATAC
atac_lsi = pd.read_csv(sp_path+"preprocessed_data/ATAC_LSI.csv")
atac_lsi['barcode'] = atac_lsi['Unnamed: 0'].str.split('#').str[1]
atac_lsi.set_index('barcode', inplace=True)
atac_lsi = atac_lsi.drop(columns = ['Unnamed: 0'])
atac_adata = ad.AnnData(atac_lsi)
atac_adata.obsm['spatial'] = rna[atac_adata.obs_names].obsm['spatial']
atac_adata.obsm['X_lsi'] = atac_adata.X
atac_adata.write_h5ad(sp_path+'ATAC_preprocessed.h5ad')
/tmp/ipykernel_1199733/3282441018.py:6: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour.
atac_adata = ad.AnnData(atac_lsi)
[ ]:
# H3K27me3
me3_lsi = pd.read_csv(sp_path+"preprocessed_data/H3K27me3_LSI.csv")
me3_lsi['barcode'] = me3_lsi['Unnamed: 0'].str.split('#').str[1]
me3_lsi.set_index('barcode', inplace=True)
me3_lsi = me3_lsi.drop(columns = ['Unnamed: 0'])
me3_adata = ad.AnnData(me3_lsi)
me3_adata.obsm['spatial'] = rna[me3_adata.obs_names].obsm['spatial']
me3_adata.obsm['X_lsi'] = me3_adata.X
me3_adata.write_h5ad(sp_path+'H3K27me3_preprocessed.h5ad')
/tmp/ipykernel_1199733/2960487857.py:6: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour.
me3_adata = ad.AnnData(me3_lsi)
[ ]:
# H3K27ac
ac_lsi = pd.read_csv(sp_path+"preprocessed_data/H3K27ac_LSI.csv")
ac_lsi['barcode'] = ac_lsi['Unnamed: 0'].str.split('#').str[1]
ac_lsi.set_index('barcode', inplace=True)
ac_lsi = ac_lsi.drop(columns = ['Unnamed: 0'])
ac_adata = ad.AnnData(ac_lsi)
ac_adata.obsm['spatial'] = rna[ac_adata.obs_names].obsm['spatial']
ac_adata.obsm['X_lsi'] = ac_adata.X
ac_adata.write_h5ad(sp_path+'H3K27ac_preprocessed.h5ad')
/tmp/ipykernel_1199733/117465859.py:6: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour.
ac_adata = ad.AnnData(ac_lsi)
[6]:
# load preprocessed ATAC, histone
atac_adata = ad.read_h5ad(sp_path+'ATAC_preprocessed.h5ad')
histme3_adata = ad.read_h5ad(sp_path+'H3K27me3_preprocessed.h5ad')
histac_adata = ad.read_h5ad(sp_path+'H3K27ac_preprocessed.h5ad')
Preprocessing
Preprocess each modality’s data and store in a dictionary output for SCIGMA
[8]:
# preprocess into dict
shared_cells = list(set(rna.obs_names).intersection(set(atac_adata.obs_names)).intersection(set(histac_adata.obs_names)).intersection(set(histme3_adata.obs_names)))
data_dict = preprocessing(rna[shared_cells], atac_adata[shared_cells], datatype='Multiplex',n_neighbors=18, feat_neighbors=6)
Preprocessing anndatas
/users/schang59/anaconda/glue/lib/python3.8/site-packages/scanpy/preprocessing/_simple.py:251: ImplicitModificationWarning: Trying to modify attribute `.var` of view, initializing view as actual.
adata.var['n_cells'] = number
/users/schang59/anaconda/glue/lib/python3.8/site-packages/anndata/_core/anndata.py:1830: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
/users/schang59/anaconda/glue/lib/python3.8/site-packages/anndata/_core/anndata.py:1830: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
/users/schang59/anaconda/glue/lib/python3.8/site-packages/anndata/_core/anndata.py:1830: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
/users/schang59/anaconda/glue/lib/python3.8/site-packages/anndata/_core/anndata.py:1830: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
Constructing spatial graphs
Constructing feature graphs
Feature graph 1
Feature graph 1 finished
Feature graph 2
Feature graph 2 finished
[9]:
# histone modalities
shared_cells = list(set(data_dict['adata_omics1'].obs_names).intersection(set(data_dict['adata_omics2'].obs_names)).intersection(set(histac_adata.obs_names)).intersection(set(histme3_adata.obs_names)))
histac_adata = histac_adata[shared_cells]
histme3_adata = histme3_adata[shared_cells]
# feat
print("processing feat1")
histac_adata.obsm['feat'] = np.nan_to_num(histac_adata.obsm['X_lsi'],nan=0).copy()
print("processing feat2")
histme3_adata = histme3_adata[histac_adata.obs_names]
histme3_adata.obsm['feat'] = np.nan_to_num(histme3_adata.obsm['X_lsi'],nan=0).copy()
# spatial graph
print("processing spatial")
cell_position_omics2 = histac_adata.obsm['spatial']
adj_omics2 = build_network(cell_position_omics2, n_neighbors=18)
histac_adata.uns['adj_spatial'] = adj_omics2
cell_position_omics2 = histme3_adata.obsm['spatial']
adj_omics2 = build_network(cell_position_omics2, n_neighbors=18)
histme3_adata.uns['adj_spatial'] = adj_omics2
# feature graph
print("processing feature spatial")
feature_graph_omics1, feature_graph_omics2 = feature_graph(histac_adata, histme3_adata, k=6)
histac_adata.obsm['adj_feature'], histme3_adata.obsm['adj_feature'] = feature_graph_omics1, feature_graph_omics2
processing feat1
processing feat2
processing spatial
processing feature spatial
Feature graph 1
Feature graph 1 finished
Feature graph 2
Feature graph 2 finished
[10]:
# protein modality
shared_cells = list(set(data_dict['adata_omics1'].obs_names).intersection(set(data_dict['adata_omics2'].obs_names)).intersection(set(histac_adata.obs_names)).intersection(set(histme3_adata.obs_names)))
protein = clr_normalize_each_cell(protein[shared_cells])
protein.layers['logcounts'] = protein.X
sc.pp.scale(protein)
sc.pp.pca(protein, n_comps=30)
protein.obsm['feat'] = protein.obsm['X_pca'].copy()
# spatial graph
cell_position_omics = protein.obsm['spatial']
adj_omics = build_network(cell_position_omics, n_neighbors=18)
protein.uns['adj_spatial'] = adj_omics
# feature graph
feature_graph_omics=kneighbors_graph(protein.obsm['feat'], 6, mode="connectivity", metric="correlation", include_self=False, n_jobs=-1)
protein.obsm['adj_feature'] = feature_graph_omics
[11]:
data_dict['adata_omics3'] = histac_adata[shared_cells]
data_dict['adata_omics4'] = histme3_adata[shared_cells]
data_dict['adata_omics5'] = protein[shared_cells]
Visualize modality specific data
[43]:
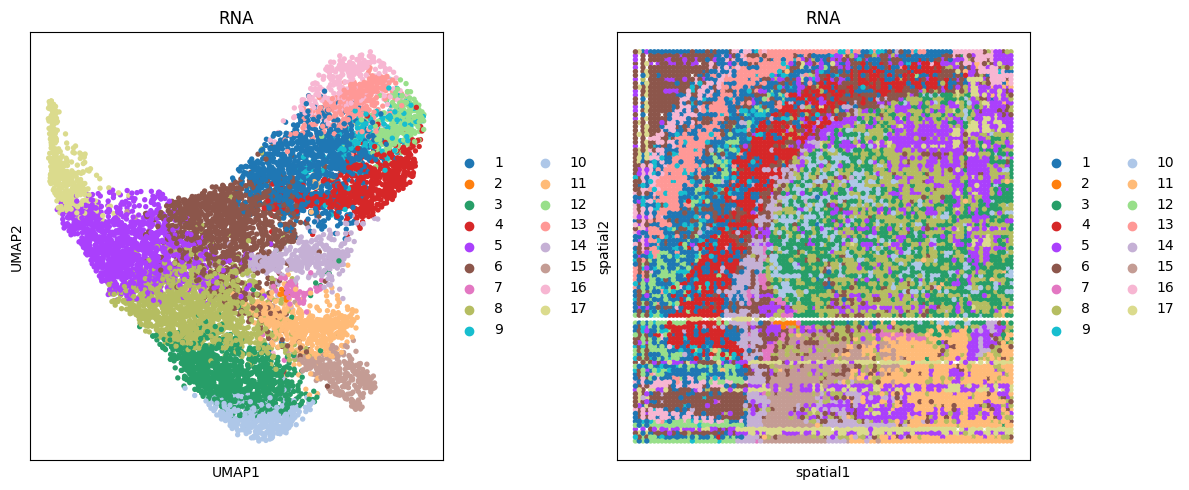
# mRNA
clustering(data_dict['adata_omics1'], key='feat', add_key='RNA', n_clusters=17, use_pca=False)
fig, ax_list = plt.subplots(1, 2, figsize=(12, 5))
sc.pp.neighbors(data_dict['adata_omics1'], use_rep='feat', n_neighbors=10)
sc.tl.umap(data_dict['adata_omics1'])
sc.pl.umap(data_dict['adata_omics1'], color='RNA', ax=ax_list[0], title='RNA', s=60, show=False)
sc.pl.embedding(data_dict['adata_omics1'], basis='spatial', color='RNA', ax=ax_list[1], title='RNA', s=60, show=False)
plt.tight_layout(w_pad=0.3)
plt.show()
fitting ...
|======================================================================| 100%
[49]:
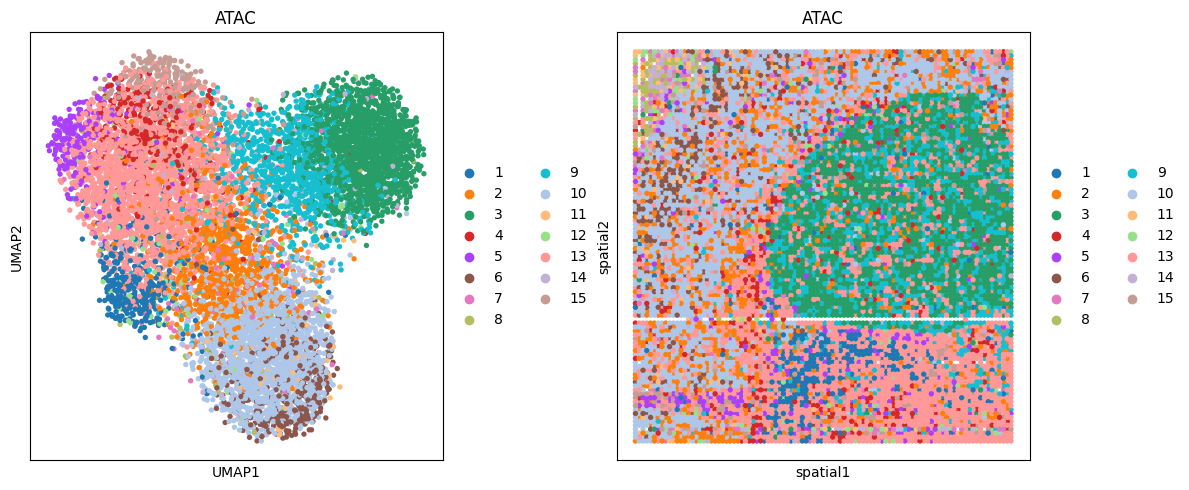
# ATAC
clustering(data_dict['adata_omics2'], key='X_lsi', add_key='ATAC', n_clusters=15,use_pca=False)
fig, ax_list = plt.subplots(1, 2, figsize=(12, 5))
sc.pp.neighbors(data_dict['adata_omics2'], use_rep='feat', n_neighbors=10)
sc.tl.umap(data_dict['adata_omics2'])
sc.pl.umap(data_dict['adata_omics2'], color='ATAC', ax=ax_list[0], title='ATAC', s=60, show=False)
sc.pl.embedding(data_dict['adata_omics2'], basis='spatial', color='ATAC', ax=ax_list[1], title='ATAC', s=60, show=False)
plt.tight_layout(w_pad=0.3)
plt.show()
fitting ...
|======================================================================| 100%
[45]:
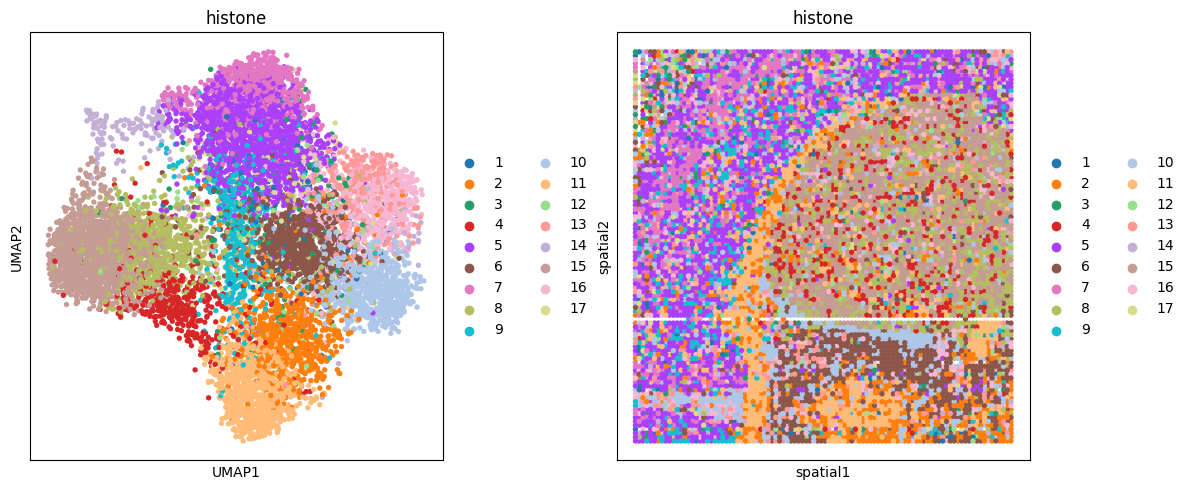
# histone (ac)
clustering(data_dict['adata_omics3'], key='feat', add_key='histone', n_clusters=17, use_pca=False)
fig, ax_list = plt.subplots(1, 2, figsize=(12, 5))
sc.pp.neighbors(data_dict['adata_omics3'], use_rep='feat', n_neighbors=10)
sc.tl.umap(data_dict['adata_omics3'])
sc.pl.umap(data_dict['adata_omics3'], color='histone', ax=ax_list[0], title='histone', s=60, show=False)
sc.pl.embedding(data_dict['adata_omics3'], basis='spatial', color='histone', ax=ax_list[1], title='histone', s=60, show=False)
plt.tight_layout(w_pad=0.3)
plt.show()
fitting ...
|======================================================================| 100%
[46]:
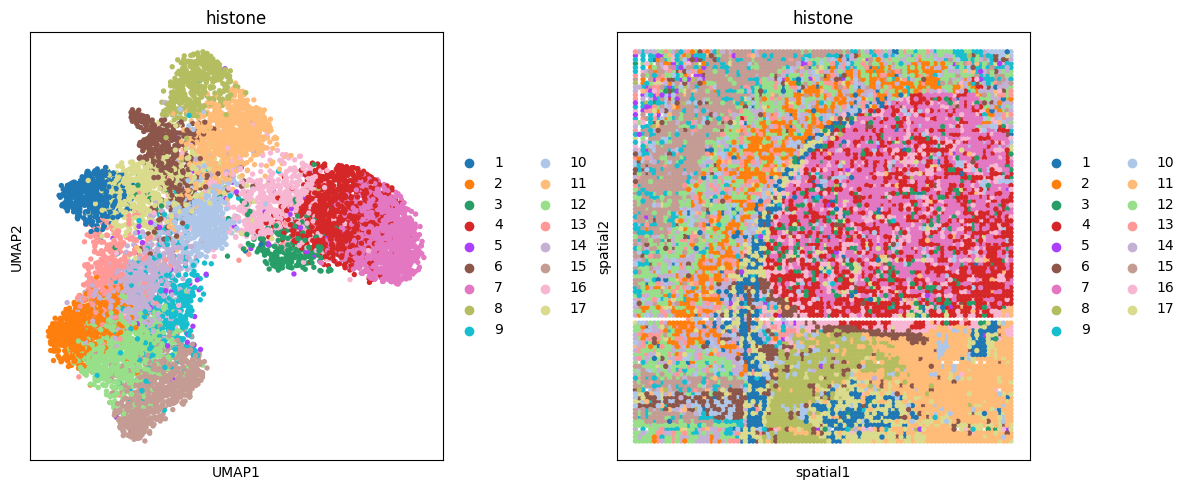
# histone(me3)
clustering(data_dict['adata_omics4'], key='feat', add_key='histone', n_clusters=17,use_pca=False)
fig, ax_list = plt.subplots(1, 2, figsize=(12, 5))
sc.pp.neighbors(data_dict['adata_omics4'], use_rep='feat', n_neighbors=10)
sc.tl.umap(data_dict['adata_omics4'])
sc.pl.umap(data_dict['adata_omics4'], color='histone', ax=ax_list[0], title='histone', s=60, show=False)
sc.pl.embedding(data_dict['adata_omics4'], basis='spatial', color='histone', ax=ax_list[1], title='histone', s=60, show=False)
plt.tight_layout(w_pad=0.3)
plt.show()
fitting ...
|======================================================================| 100%
Exception ignored from cffi callback <function _processevents at 0x7f05fb6ff790>:
Traceback (most recent call last):
File "/users/schang59/anaconda/glue/lib/python3.8/site-packages/rpy2/rinterface_lib/callbacks.py", line 277, in _processevents
try:
KeyboardInterrupt:
[47]:
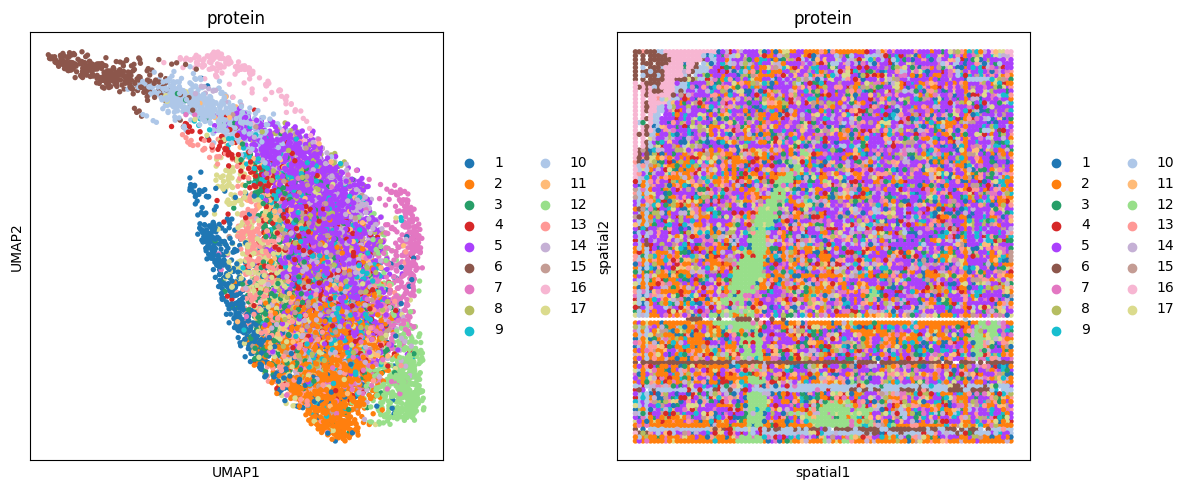
# protein
clustering(data_dict['adata_omics5'], key='feat', add_key='protein', n_clusters=17,use_pca=False)
fig, ax_list = plt.subplots(1, 2, figsize=(12, 5))
sc.pp.neighbors(data_dict['adata_omics5'], use_rep='feat', n_neighbors=10)
sc.tl.umap(data_dict['adata_omics5'])
sc.pl.umap(data_dict['adata_omics5'], color='protein', ax=ax_list[0], title='protein', s=60, show=False)
sc.pl.embedding(data_dict['adata_omics5'], basis='spatial', color='protein', ax=ax_list[1], title='protein', s=60, show=False)
plt.tight_layout(w_pad=0.3)
plt.show()
fitting ...
| | 0%
Exception ignored from cffi callback <function _processevents at 0x7f05fb6ff790>:
Traceback (most recent call last):
File "/users/schang59/anaconda/glue/lib/python3.8/site-packages/rpy2/rinterface_lib/callbacks.py", line 277, in _processevents
try:
KeyboardInterrupt:
|======================================================================| 100%
Model Training
[134]:
seed = 2025
contrastive_weights = {'adata_omics1': 1e-1, 'adata_omics2': 1e-1, 'adata_omics3': 1e-1, 'adata_omics4': 1e-1, 'adata_omics5': 1e-1}
recon_weights = {'adata_omics1': 1, 'adata_omics2': 1, 'adata_omics3': 1, 'adata_omics4': 1, 'adata_omics5': 1}
model = SCIGMA_multi(data_dict, seed_num=seed, dim_output=40, device=torch.device('cuda:0'), clr_weight=0.3, batch_size=3000, learning_rate=1e-3, contrastive_weights=contrastive_weights, recon_weights=recon_weights, connectivities=None)
Creating adjacency matrices
Model ready for training!
[ ]:
output = model.train(1500)
[136]:
adata_combined = data_dict['adata_omics1'].copy()
adata_combined.uns['emb_latent'] = output['emb_latent']
adata_combined.uns['emb_recon'] = output['emb_recon']
adata_combined.obsm['emb_combined'] = output['emb_latent_combined']
adata_combined.obs['invtau'] = output['invtau']
adata_combined.obs['tau'] = output['tau']
adata_combined.uns['attention'] = output['attention']
/users/schang59/anaconda/glue/lib/python3.8/site-packages/anndata/_core/anndata.py:1830: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
Spatial Domain Detection
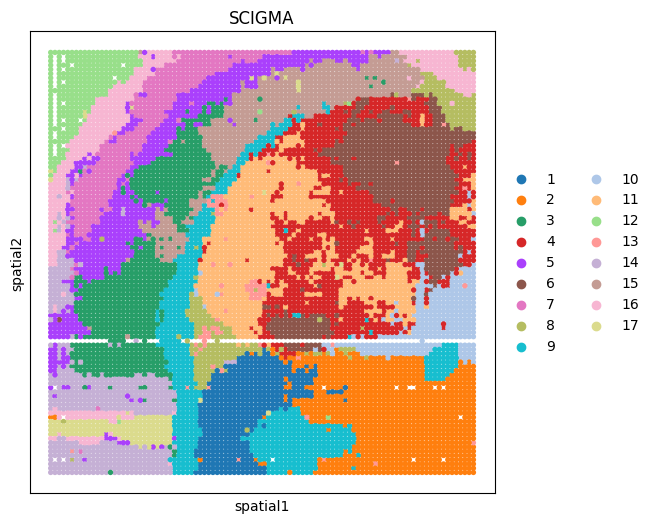
[137]:
# we use mcluster as clustering tool by default.
tool = 'mclust' # mclust, leiden, and louvain
# clustering
if tool == 'mclust':
clustering(adata_combined, key='emb_combined', add_key='SCIGMA', n_clusters=17, method=tool, use_pca=False)
elif tool in ['leiden', 'louvain']:
clustering(adata_combined, key='emb_combined', add_key='SCIGMA', n_clusters=17, method=tool, start=0.1, end=2.0, increment=0.01)
fitting ...
|======================================================================| 100%
[138]:
# visualization
import scanpy as sc
fig, ax_list = plt.subplots(1, 1, figsize=(6, 6))
sc.pl.embedding(adata_combined, basis='spatial', color='SCIGMA', ax=ax_list, title='SCIGMA', s=60, show=False)
plt.gca()
plt.show()
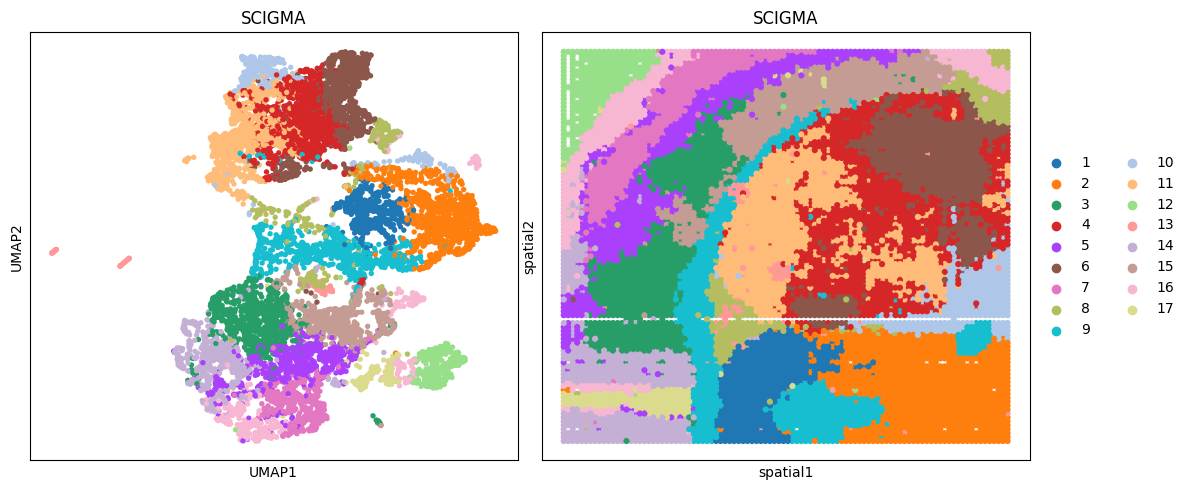
[139]:
# visualization
fig, ax_list = plt.subplots(1, 2, figsize=(12, 5))
sc.pp.neighbors(adata_combined, use_rep='emb_combined', n_neighbors=10)
sc.tl.umap(adata_combined)
sc.pl.umap(adata_combined, color='SCIGMA', ax=ax_list[0], title='SCIGMA', s=60, show=False)
sc.pl.embedding(adata_combined, basis='spatial', color='SCIGMA', ax=ax_list[1], title='SCIGMA', s=80, show=False)
ax_list[0].get_legend().remove()
plt.tight_layout(w_pad=0.3)
plt.show()
Uncertainty Visualization
[ ]:
# uncertainty visualization
sc.pl.embedding(adata_combined,
basis='spatial',
color=['invtau'],
title=['Uncertainty'], s=50, show=False)
plt.gca()
plt.show()
[ ]: